The simulation log file is a file with the same name as the input file and the extension *.log.
It is located in the output folder of the simulation.
It is necessary for the support team to debug issues with the simulation, thus it should always be attached to a support request.
Please also include additional debug information in case the log file shall be used for support, refer to How to add additional debug information to the LOG file.
How to add additional debug information to the LOG file¶
Within the nextnanomat options Options: Expert settings some flags can be found which add additional information to the log file.
Please check the following flags and re-run the simulation so the log file will include this information:
‘Include simulation time in log file’
‘Include memory usage in log file (units: MB)’
‘Include additional debug information in log file’
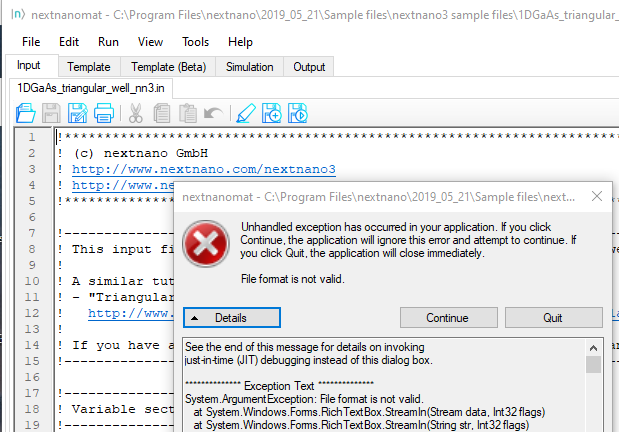
ERROR when loading input file “File format is not valid”¶
A beta feature in a recent Windows 10 version (1803) causes the following error message when starting nextnanomat:
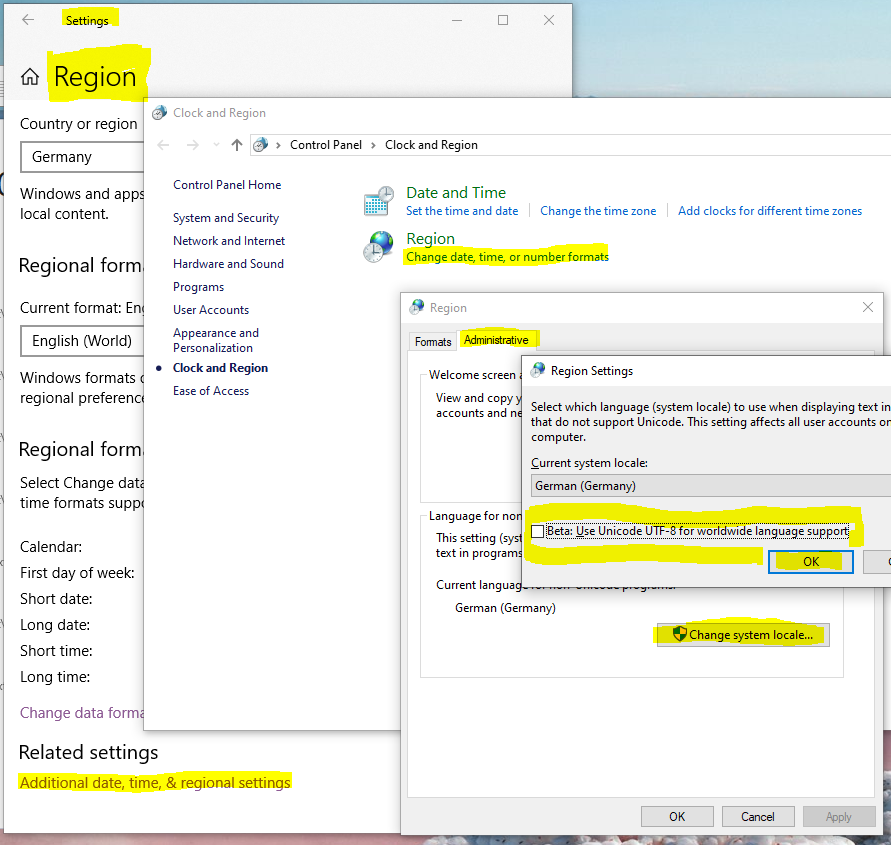
You can disable this feature in the Region’s settings
of Windows (screenshot see Figure 12).
Select Additionaldate,time,®ionalsetting
Then Changedate,time,ornumberformats
Go the the AdministrativeTab
Click Changesystemlocale...
And finally you can un-select that Beta-Feature.
Figure 12 How to change Windows 10 region settings.¶
Error while starting simulation (“The specified executable is not a valid application for this OS platform.”)¶
If you get this error, you are executing an executable that is not suited for this operating system.
There are two possible reasons:
You are executing a 64-bit version on a 32-bit operating system.
Solution: Specify the location of the 32-bit nextnano++ or nextnano³ executables.
Tools ==> Options… ==> Simulation ==>nextnano GmbH executable file ==>32bit.exe
You are executing the Intel or Microsoft 64-bit executable that has not been compiled for your rather old CPU.
Our 64-bit executables use more advanced CPU instructions compared to our 32-bit executables.
Solution: Use the 32-bit nextnano++ executable that has been compiled with the Microsoft compiler.
Tools ==> Options… ==> Simulation ==>nextnano++ executable file ==>nextnano++_Microsoft_32bit.exe
Sometimes the displayed error message does not contain any content apart from ERROR.
You can share information about your CPU with us using the menu
Help==>GenerateSystemSnapshotforSupport and then send us the generated file nextnano_SystemSnapshot.txt.
My Quantum-Current-Poisson calculation fails to converge.
How can I avoid this problem?
Answer
The most common errors are the following:
The maximum number of iterations has been chosen too small (run{ }==>outer_iteration{}==>iterations) and the calculation just needs some more iterations.
The number of electron or hole eigenstates has been chosen too small (e.g. quantum{ }==>region{}==>Gamma{}==>num_ev).
Check the occupations in the files wf_spectrum_"namequantumregion"_"namequantummodel".dat.
If the occupation does not drop from the ground state to the highest excited state by several orders of magnitude,
you need to calculate more states (= more possibilities to fill in charges).
The under-relaxation parameter has been chosen too large (run{ }==>outer_iteration{}==>alpha_current).
Check whether the solution oscillates (residuals do not drop continuously but increase in some iterations).
Try to decrease the under-relaxation parameter in order to damp the oscillations.
The minimum charge density in the current equation has been chosen too small (currents{ }==>minimum_density).
Try to increase the minimum charge density to improve conditioning of the current equation.
In fact, the current equation is solved with the charge density = max( real density, minimum_density).