Absorption of a GaAs spherical quantum dot¶
Section author: Naoki Mitsui
This tutorial calculates the optical absorption spectrum of a GaAs spherical quantum dot with infinite barriers. We will see which output file we should refer to in order to understand the absorption spectrum.
Also, the formula used for the absorption calculation is presented. For the detailed scheme of the calculation of the optical matrix elements and absorption spectrum, please see our 1D optics tutorial: Optical absorption for interband and intersubband transitions
Input file:
3Dspherical_infinite_dot_GaAs_intra_nnp.in
3Dspherical_infinite_dot_GaAs_inter_nnp.in
Structure¶
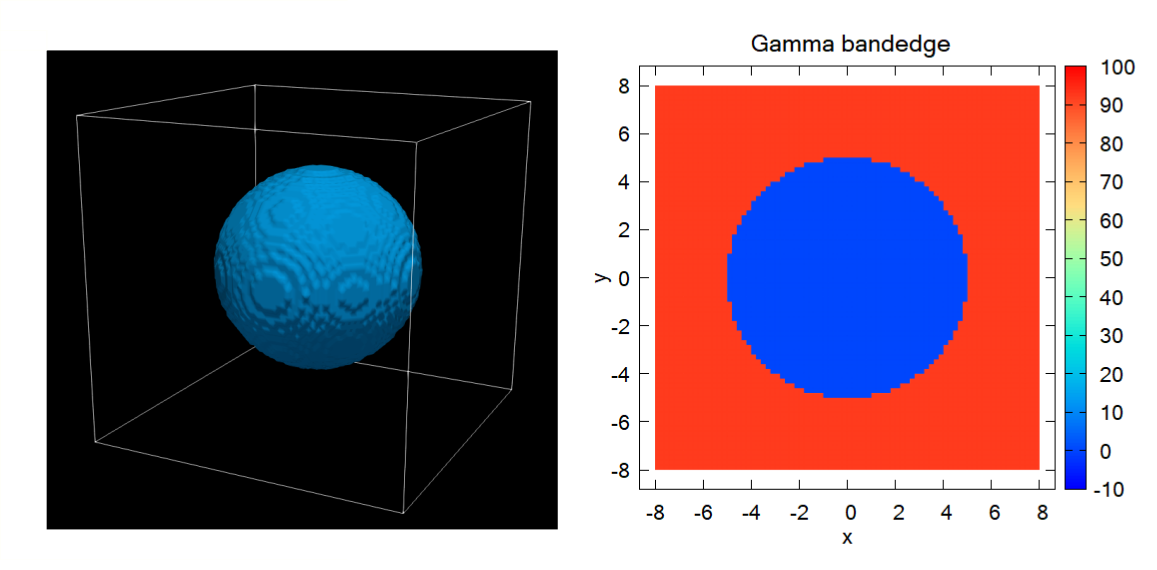
Figure 2.5.12.77 Left: GaAs region as a spherical quantum dot. Right: Slice of the Gamma band edge along \(z=0\).¶
The above figures show the Gamma band edge of the spherical GaAs region and the barrier region.
We model the infinite barrier by assigning 100 eV for the band edge of AlAs barrier region from database{} section.
Please see the input file for the details.
The parameters used in this simulation are as follows.
Property |
Symbol |
Value [unit] |
|---|---|---|
quantum dot radius |
\(R\) |
5 [nm] |
barrier height |
\(E_b\) |
92 [eV] |
effective electron mass |
\(m_e\) |
0.0665 |
refractive index |
\(n_r\) |
3.3 |
doping concentation (n-type) |
\(N_D\) |
8\(\cdot\)1018 [cm-3] |
linewidth (FWHM) |
\(\Gamma\) |
0.01 [eV] |
temperature |
\(T\) |
300 [K] |
Scheme¶
The run{} section is specified as follows:
run{
poisson{}
quantum{}
optics{}
}
Then the simulation follows these steps:
Poisson equation is solved with the setting specified in the poisson{} (optional) section.
“Schrödinger” equation is solved with the setting specified in the quantum{ } section.
“Schrödinger” equation is solved again with the setting specified in the optics{ } section and optical properties are calculated.
Note
If
quantum_poisson{}is specified instead ofquantum{}, Poisson and Schrödinger equations are solved self-consistently.optics{}requires that kp8 model is used in the quantum region specified inquantum{}.In this tutorial the kp parameters are adjusted so that the conduction and valence bands are decoupled from each other. Thus the single-band Schrödinger equations are solved effectively by the kp solver.
The optical absorption accompanied by the excitation of charge carriers (state \(n\to m\)) in a condensed matter is calculated on the basis of Fermi’s golden rule [ChuangOpto1995] in the dimenstion of (length)-1:
where
\(E_{n}\) is the energy of eigenstate \(n\). The first sum runs over the pair of states where \(E_{n}>E_{m}\).
\(f_n\) is the occupation of eigestate \(n\).
\(\vec{\epsilon}\) is the optical polarization vector defined in optics{ region{} }.
\(\vec{\pi}=\vec{p}+\frac{1}{4m_0c^2}(\sigma\times\nabla V)\) where \(\vec{p}\) is the canonical momentum operator and \(\frac{1}{4m_0c^2}(\sigma\times\nabla V)\) is the contribution of spin-orbit interaction.
\(\vec{\pi}_{nm}=\langle n|\vec{\pi}|m\rangle\).
we call \(\vec{\epsilon}\cdot\vec{\pi}_{nm}\) as the optical matrix elements.
- \(\mathcal{L}(E_n-E_m-\hbar\omega)\) is the energy broadening function:
When
energy_broadening_lorentzianis specified in optics{ region{} },\(\mathcal{L}(E_n-E_m-\hbar\omega)=\frac{1}{\pi}\frac{\Gamma/2}{(E_n-E_m-\hbar\omega)+(\Gamma/2)^2}\)
where \(\Gamma\) is the FWHM defined by
energy_broadening_lorentzian.When
energy_broadening_gaussianis specified in optics{ region{} },\(\mathcal{L}(E_n-E_m-\hbar\omega)=\frac{1}{\sqrt{2\pi}\sigma}\exp{\big(-\frac{(E_n-E_m-\hbar\omega)^2}{2\sigma^2}\big)}\)
where
energy_broadening_lorentziandefines the FWMH \(\Gamma=2\sqrt{\ln 2}\cdot\sigma\)When neither
energy_broadening_lorentziannorenergy_broadening_gaussianis specified inoptics{ region{} }, \(\mathcal{L}\) is replace by the delta function \(\delta(E_n-E_m-\hbar\omega)\).It is also possible to include both Lorentzian and Gaussian broadening (Voigt profile).
The detailed calculation scheme of the optical matrix elements \(\vec{\epsilon}\cdot\vec{\pi}_{nm}\) is described in Optical absorption for interband and intersubband transitions. In 3D simulation we don’t have the k-summation like 1D and 2D cases.
Results¶
Absorption¶
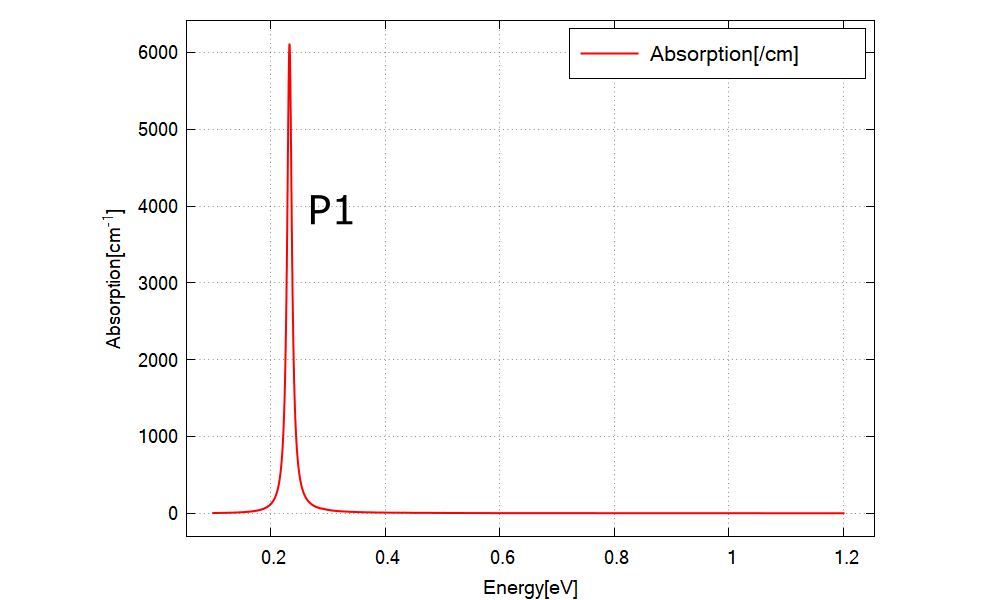
Figure 2.5.12.78 Calculated absorption spectrum \(\alpha(\vec{\epsilon}, E)\) for \(\vec{\epsilon}=\hat{x}\).¶
Figure 2.5.12.78 shows the calculated \(\alpha(\vec{\epsilon}, E)\) specified in \Optics\absorption_~.dat for x-polarization. The absorptions for y- and z-polarization are identical to this graph due to the rotational symmetry. We have one peak at around 0.23 eV (P1). These results can be understood from the output data explained below.
Note
When we use the realistic k.p paramters, \(\alpha(\vec{\epsilon}, E)\) for each polarization would no more be identical in general. This is because the eigenstates above the conduction band edge can have the component of valence band Bloch functions (band-mixing).
They are identical in this tutorial since the single-band model is emulated.
Eigenvalues, transition energies, and occupations¶
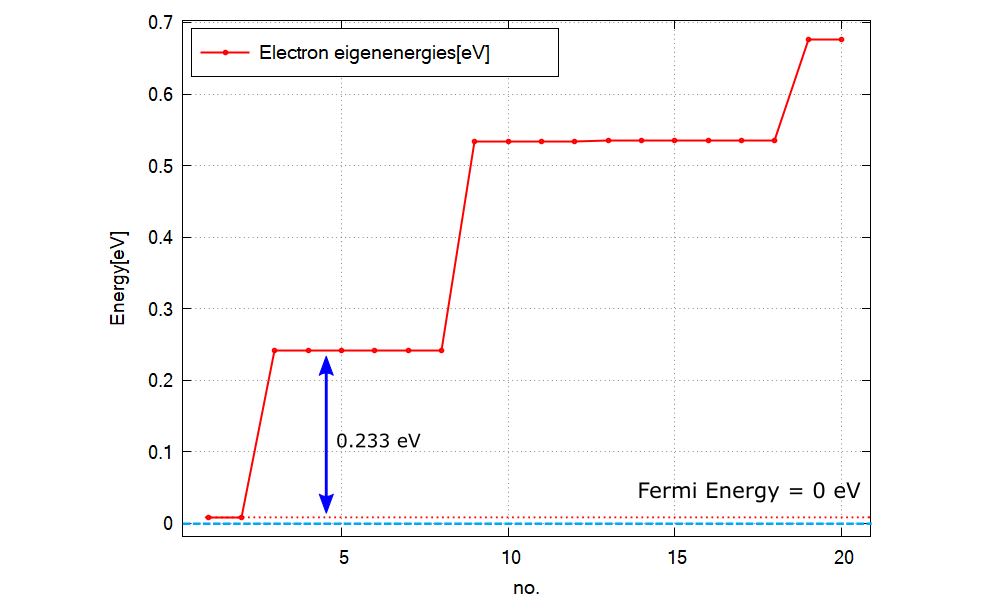
Figure 2.5.12.79 Calculated energy spectrum and Fermi energy (=0 eV).¶
Figure 2.5.12.79 shows the calculated energy eigenvalues specified in \Quantum\energy_spectrum_~.dat.
Please note that the output in Quantum\ counts the eigenstates with different spins individually when k.p model is used, while they are counted jointly in Optics\.
Comparing the excitation energy of other upper states to \(k_BT\simeq 0.026\) eV at \(T=300\) K, we can expect the occupation probability of each excited state is almost 0 and only the ground states have the non-zero occupation. Thus the optical transition will occur only from the ground states in this case.
We can see the peak energy of P1 in Figure 2.5.12.78 corresponds to the transition energy from the ground states (no. 1 and 2) to the 1st excited states (no. 3,4,5,6,7 and 8).
Note
The eigenstates with different spins are counted individually in Quantum\ when k.p model is used, while they are counted jointly in Optics\.
For example, the two ground states counted as no.1 and 2 in Figure 2.5.12.79 due to spin are put together as one eigenstate in Optics\.
From the above data of eigenvalues, we could see which pair of states contributes to the peak in the absorption spectrum Figure 2.5.12.78. In order to understand why some pairs of states don’t appear as peaks, we will see the output data for \(|\vec{\epsilon}\cdot\vec{\pi}_{nm}|^2\) next.
Transition intensity (Momentum matrix element)¶
One of the key element for the calculation of optical absorption is the transition intensity
which has the dimension of energy [eV].
The intensity \(\big(T_{nm}(\vec{\epsilon})\big)\) for each pair of states \((n,m)\) is specified in Optics\transitions_~.txt. These intensities whose “From” states are the ground state are shown here for x-polarization. We can also check the transition energy of each pair of states.
Energy[eV] From To Intensity_k0[eV] 1/Radiative_Rate[s]
0.233098 1 2 2.02882 4.43013e-08
0.233098 1 3 2.42777 3.70214e-08
0.233098 1 4 2.30413 3.90079e-08
The transtions from 1 to 5~10 are zero and these pairs of states don’t contribute to the absorption (They are omitted here since Intensity_k0 are too small).
Eigenstates¶
The probability distribution of eigenfunctions \(|\psi(\textbf{r})|^2\) is output in Quantum\probabilities_~.vtr. The amplitude of the envelope function on each Bloch function \(|S\rangle,|X\rangle,|Y\rangle,|Z\rangle\) can be found in Quantum\amplitudes_~_SXYZ.vtr.
Here the probability distribution of eigenfunctions calculated by single-band model are shown.

Figure 2.5.12.80 |wave function|\(^2\) of the ground state. (s orbital, not degenerated.)¶

Figure 2.5.12.81 |wave function|\(^2\) of the 1st excited state. (3 times degenerated, p orbital)¶
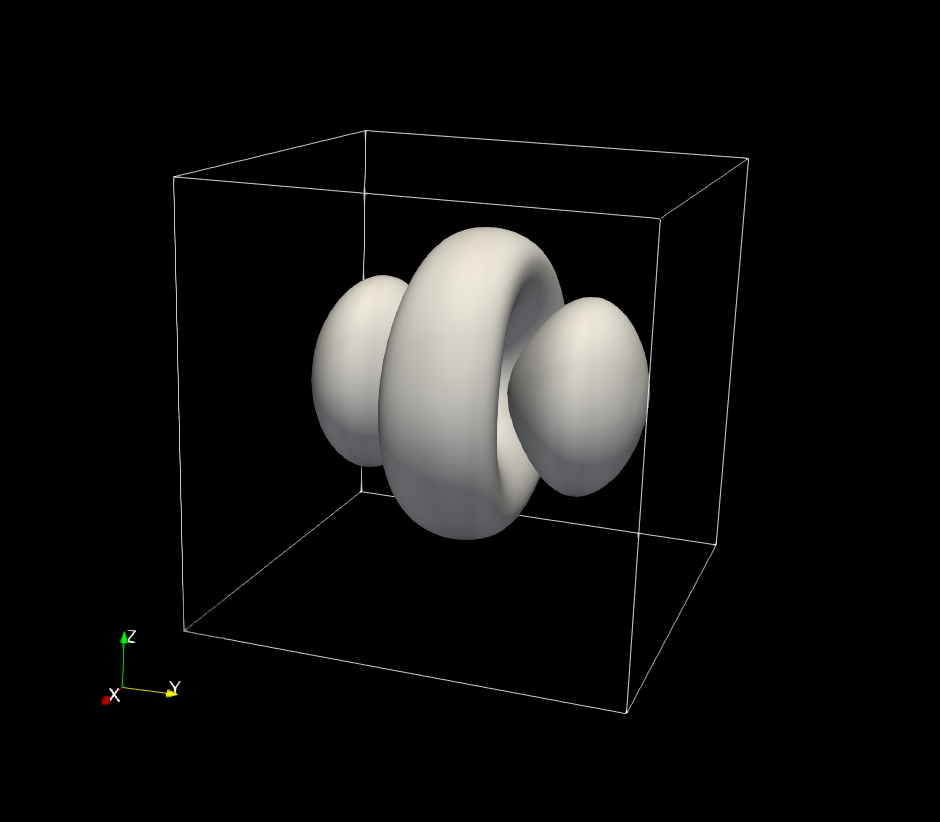
Figure 2.5.12.82 |wave function|\(^2\) of the 2nd excited state. (5 times degenerated, d orbital)¶

Figure 2.5.12.83 |wave function|\(^2\) of the 2nd excited state. (d orbital)¶
|wave function|\(^2\) of the energy eigenstates calculated by the single-band model. The contours at the value of \(|\psi(\textbf{r})|^2=0.001\) are shown.
Last update: nn/nn/nnnn