nextnano3 - Tutorial
next generation 3D nano device simulator
3D Tutorial
Hole energy levels of an "artificial atom" - Spherical Si Quantum Dot
(6-band k.p)
Author:
Stefan Birner
-> 3DsphericSiQD_d5nm_6bandkp_nn3.in / *_nnp.in -
spherical Si QD with diameter 5 nm -
input file for the nextnano3 and nextnano++ software
These input files are included in the latest version.
Hole energy levels of an "artificial atom" - Spherical Si Quantum Dot
(6-band k.p)
-> 3DsphericSiQD_d5nm_6bandkp_nn3.in -
spherical Si QD with diameter 5 nm
Here, we want to calculate the energy spectrum of a
spherical Si
quantum dot of radius 2.5 nm.
We assume that the barriers at the QD boundaries are infinite.
The potential inside the QD is assumed to be 0 eV.
We use a grid resolution of 0.25 nm.
We solve the 6-band k.p Schrödinger equation for the hole eigenstates.
The following 6-band k.p parameters are used:
L = -6.8
!
M = -4.43 !
N = -8.61 !
Deltaso = 0.044 eV
6x6kp-parameters = -6.8d0 -4.43d0 -8.61d0
! L, M, N [hbar^2/2m]
0.044d0
! Deltasplit-off [eV] spin-orbit splitting energy
gamma1 = 4.22
gamma2 = 0.395
gamma3 = 1.435
The following figure shows the hole eigenenergy spectrum of the Si QD
(diameter = 5 nm) calculated with a 6-band k.p Hamiltonian.
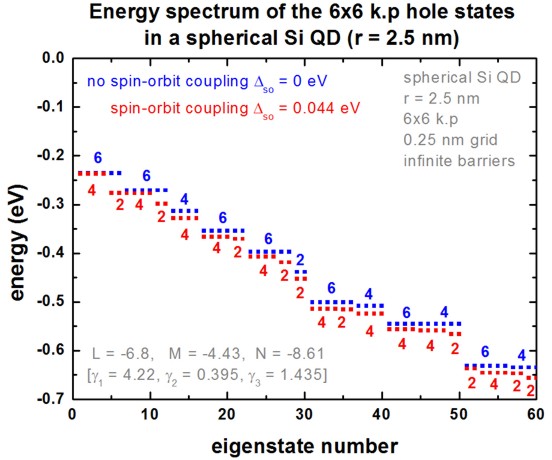
For comparison, we also display the energy spectrum where we assumed
zero spin-orbit splitting energy.
In this case there is a six-fold symmetry.
Spin-orbit splitting reduces this degeneracy to 4
and 2.
In general, each state is two-fold degenerate due to spin.
(Note: We used a cuboidal shaped quantum cluster although we could
have used a spherical quantum cluster to reduce the size of the 6-band
k.p Hamiltonian matrix that has to be solved for the eigenstates.
If m grid points can be excluded from the quantum region due to an
optimal choice of quantum region shape, the dimension N of the 6-band k.p
matrix is reduced by N-6m.)
Following the paper of [Burdov], one can calculate the ground state energy
for this particular system from the L and M parameters:
E1 = - hbar2 pi2 / ( 2
mh R2 ) = -0.314 eV (using mh
= 0.192 m0 as [Burdov] where he uses incorrect k.p parameters:
In his definition L must be -5.8 and M = -3.43.)
E1 = - hbar2 pi2
/ ( 2 mh R2 ) = -0.254 eV (using
mh = 0.237 m0 as V.A. Belyakov, V.A. Burdov, J. Phys.:
Condens. Matter 20, 025213 (2008))
The latter is in much better agreement to our calculations.
mh is given by:
mh = 3 m0 / ( L + 2 M
) = 3 m0 / ( -6.8 + 2 * (-4.43)) = -0.192 m0
[Burdov]
mh = 3 m0 / ( (L+1) + 2 (M+1)) = 3 m0 / (
-5.8 + 2 * (-3.43)) = -0.237 m0 [Belyakov/Burdov]
The latter definition is consistent to our implementation of the k.p
Hamiltonian.
The discrepancy of these equations arises because there are two different
definitions of the L,M parameters available in the literature.
More details...
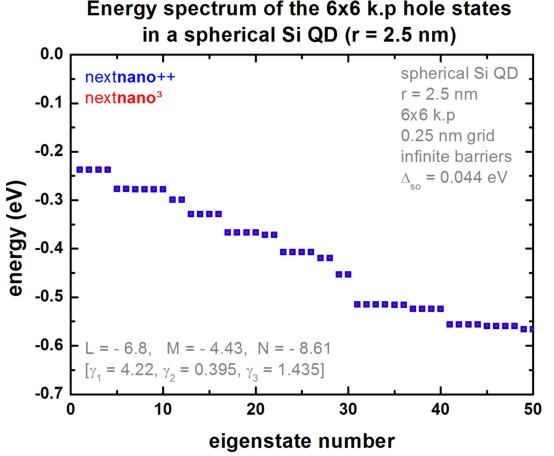
nextnano³ vs. nextnano++ comparison
The following figure compares the nextnano³ results with the nextnano++
results.
The results of both simulators are in excellent agreement.
Additional comment for experts
For this particular geometry, the eigenvalues are highly degenerate, not only
due to spin, but also due to geometry.
This might cause problems for certain eigenvalue solvers as they might miss some
of these degenerate eigenvalues.
So the software should be used with care.
In our case, the 'chearn' eigenvalue
solver (Arnoldi method that uses Chebyshev polynomials as preconditioner) missed
some degenerate eigenvalues. So probably one has to adjust some eigenvalue
solver parameters to increase the accuracy.
For this reason it is of great advantage if any numerical software has
redundancy in terms of several eigensolvers where one can choose from in order
to check results for consistency and accuracy, as well as performance. |