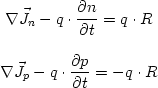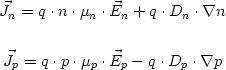1D simulation
Evaluating the density integration
The program provides three options for the calculation of the density when
performing a 1D k.p calculation. For
this, only k values up to a maximum value of k have to be considered. All
values that are larger are lying energetically very far away from the Fermi
level so that their contributions to the density can be neglected.
The option will be specified in the input file under the keywords
$quantum-model-electrons/$quantum-model-holes:
Specifier method-of-brillouin-zone-integration =
...
= special-axis
In this case the Hamiltonian must be isotropic in the (kx, ky)
plane, i.e. E(kx, ky) = E(k||) where k||²
= kx² + ky². Then the integration of the density can be
reduced to a 1D integral. This is only allowed for wurtzite in the [0001]
growth direction. In all other cases the program switches to
simple-integration.
= simple-integration
The 2D Brillouin zone will be discretized and the density will be calculated
by the summation over all k points.
For zinc blende with {001} quantization
direction only the irreducible part of the Brillouin zone is being discretized
(up to maximum values of k), correspong to about ~1/8 of the total
number of k points. In all other cases the whole Brillouin zone is
being discretized (up to maximum values of k).
= gen-dos
The evaluation of the density is done by integration over the density of
states (DOS).
For zinc blende with {001} quantization direction only the irreducible
part of the Brillouin zone is being discretized (up to maximum values of k),
correspong to about ~1/8 of the total number of k points.
In all other cases the whole Brillouin zone is being discretized (up to
maximum values of k).
The number of discretization points has to be specified under the specifier
'num-kp-parallel' and refers to the discretization of the whole
Brillouin zone up to the maximum value of k. For the special cases special-axis
and zinc blende in {001} direction, the program is calculating the
number of actually needed points.
More details can be found
here and
here and here:
$quantum-model-electrons
$quantum-model-holes